
Delivering on the Promise of Medical AI: Can Japan’s Regulators Keep Pace? (Part 2)
May 18, 2022
R-2021-060-2E
In Part 1 of this article, I traced the lengthy process by which Japan updated its regulatory framework so as to accommodate medical devices leveraging artificial intelligence and machine learning (AI/ML). In the following, I discuss some of the structural factors that are hindering the adaptation process and propose a way forward.
Regulatory Lag in the US and Japan
The problem of regulatory lag is not unique to Japan. In fact, it took the US Food and Drug Administration almost as long to develop and institute evaluation methods suited to ML-enabled medical devices and their ever-changing performance. Let us begin by examining the FDA’s response and comparing it to Japan’s.
For regulatory purposes, the FDA distinguishes between AI-enabled medical devices using “locked” algorithms and those using continuously “adaptive” algorithms. Locked algorithms are defined as those that provide the same result each time the same input is applied to them and do not change with use, while “adaptive” algorithms analyze the data they collect and modify their own behavior accordingly. Until February 2020, the only AI-enabled medical devices the FDA had approved were those using locked algorithms.
The first clear sign of a change in this approach came in April 2019 with the FDA’s release of a discussion paper titled “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as Medical Device.” The paper, which was accompanied by a request for feedback, outlined a proposal for the regulation of AI-based medical devices using continuously changing adaptive algorithms, including the submission of a “Predetermined Change Control Plan.” The proposal was subsequently modified on the basis of extensive stakeholder feedback (including workshops), and in February 2020 the FDA announced its first market authorization of adaptive ML-enabled medical software using the new regulatory system. What this means, in effect, is that until February 2020, the FDA—just like Japan’s PMDA before the revision of the Pharmaceuticals and Medical Devices Act—was unable to approve premarket submissions of AI/ML-based medical whose performance changed with use.
A distinguishing feature of the FDA’s approach was its development of an ambitious new quality control policy under the name of Good Machine Learning Practice, or GMLP (see figure below), which calls on developers and manufacturers to practice a “culture of quality and organizational excellence” at every stage in the product life cycle: data selection and management, model training and tuning, model evaluation and validation, retraining based on real-world performance monitoring, and so forth. In essence, it attempts to regulate not just product performance per se but also the manufacturer’s quality-control efforts across the product life cycle (as through quality assurance plans and protocols for managing data and updating algorithms). This is a novel approach to regulation and qualitatively different from Japan’s.
In January 2021, the FDA issued an Artificial Intelligence/Machine Learning–Based Software as a Medical Device Action Plan. The plan promises to issue draft guidance on companies’ Predetermined Change Control Plan (a key aspect of the new regulatory framework), much as Japan’s revised Pharmaceuticals and Medical Devices Act calls for a Post-Approval Change Management Protocol. It also pledges to “strengthen FDA’s encouragement of the harmonized development of Good Machine Learning Practice (GMLP) through additional FDA participation in collaborative communities and consensus standards development efforts” and to “support regulatory science efforts on the development of methodology for the evaluation and improvement of machine learning algorithms.” In addition, the FDA joined with Health Canada and Britain’s Medicines & Healthcare Products Regulatory Agency in drafting 10 guiding principles of GMLP, which it published in October 2021.
Looking back over the FDA’s response, it is clear that US regulators struggled no less than their Japanese counterparts with the question of how to regulate the new technology represented by AI-based medical devices. In fact, Japan appears to have embarked on the process of updating its regulatory framework before the United States, as suggested by the appearance of the aforementioned PMDA report (see Part 1) in December 2017.
Nonetheless, in terms of putting such ideas into practice, the United States beat Japan to the finish line: The FDA authorized the first medical device using adaptive ML in February 2020, whereas the PMDA was unable to issue such authorizations until the following September, when the new law came into effect. The FDA has also moved with great speed to draw up GMLP standards in collaboration with overseas regulatory agencies.
The FDA’s Good Machine Learning Practice (GMLP) Concept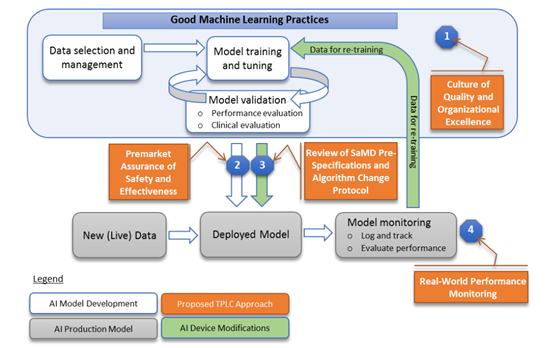
Source: Reprinted from Food and Drug Agency, “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as Medical Device,” https://www.fda.gov/files/medical%20devices/published/US-FDA-Artificial-Intelligence-and-Machine-Learning-Discussion-Paper.pdf.
Critiquing Japan’s Rulemaking Process
Having reviewed the processes by which Japan and the United States each attempted to formulate new regulatory frameworks geared to AI-based medical devices, let us examine some of the structural features that distinguished the rulemaking process in Japan.
First, it was the PMDA, an incorporated administrative agency—not the Medical Device Evaluation Division of the Ministry of Health, Labor, and Welfare—that initiated the process and set the agenda. This is quite unusual. Incorporated administrative agencies are established with the ostensible goal of separating policy planning from policy execution by keeping the former function in the ministries while transferring the latter to agencies like the PMDA. Within this framework, the basic purpose of the PMDA is to carry out premarket reviews and postmarket monitoring based on regulatory policies established by the MHLW. In this instance, however, the PMDA played the leading role in proposing and drafting a new regulatory framework.
The most likely reason for this arrangement is that, even though the MHLW has jurisdiction over the regulation of pharmaceuticals and medical devices, it lacks the capacity for policy planning in this field. Of course, the MHLW has many technical officers trained in fields like medical engineering, but when it comes to designing a whole new regulatory framework for an emerging technology, these technocrats would be hard put to know when to embark on such a project, let alone identify that technology’s novel features and the best means by which to assess them.
Within the PMDA, moreover, it was a subcommittee of the Science Board, made up of outside experts, that was responsible for the drafting of early recommendations, not any of the permanent units in charge of review or assessment methodology. Not being a national research institution, the PMDA is not equipped to conduct serious study in the field of regulatory science. In all likelihood, the agency had no choice but to rely on the expertise of scholars in the pharmaceutical or medical engineering departments of major universities.
A related issue concerns the capabilities of the officers whose task it is to review actual applications for regulatory approval. As street-level officers in a regulatory agency, PMDA reviewers are charged with conducting evaluations using clearly defined methods, procedures, and criteria. But when a new technology appears for which the rules have yet to be firmly established, they are unable to perform the evaluations assigned to them.
Perhaps the biggest issue with the current rulemaking process is the extremely limited role given to the private companies that are actually conducting cutting-edge R&D with a view to bringing innovative medical products to the market. As noted in Part 1, a certain amount of dialogue with the makers of diagnostic imaging systems did take place back in December 2016, but feedback opportunities were limited to a handful of “opinion exchanges” and hearings, and there is no indication that these exchanges had any substantive impact.
Overcoming Regulatory Lag
Regulatory lag should not occur if regulators are attuned to the latest technological developments and equipped to respond in a timely and appropriate manner. But the reality is that our regulators are beginning to find themselves unequal to the task of anticipating and responding to the challenges posed by emerging technologies, as we have seen in the case of AI-based medical devices. As technology continues to advance, industry will doubtless continue to develop innovative products and services that are difficult or impossible for regulators to evaluate using established methods. That being the case, we need to consider new policies to minimize regulatory lag with the capabilities of our regulators in mind.
If our bureaucrats lack the capacity to deliberate and draft new regulations by themselves, then we have little choice but to enlist the participation of those who have that capacity. In the case of AI-based medical devices, experts from the academic sector were brought in for that purpose. But it is industry, not academia, that is directly engaged in the development of innovative products and services incorporating AI. Surely the private companies that are developing technically innovative medical products have the most information about those products’ novel features. In the case of regenerative medicine, the development of a new regulatory framework was a collaborative process between administrative authorities, academia, and industry, with business groups and loose confederations of companies (such as the Forum for Innovative Regenerative Medicine) joining in the rulemaking process alongside the Japanese Society for Regenerative Medicine, an academic organization.
Of course, the participation of private industry in the regulatory planning process must be handled carefully. Companies that are asked to contribute their expertise will need assurances that their intellectual property rights will be protected. There must also be controls to guard against the possibility that private companies will be tempted to shape the new regulations to their own advantage, as by lowering the standard of evidence required to establish safety or efficacy. But the alternative is to adhere to the traditional approach, relying on regulators who are unable to keep pace with technological progress. It seems to me that, rather than fall into a state of permanent regulatory dysfunction, we should move decisively to develop a new approach predicated on the collaboration of industry, government, and academia, both in the design of new regulations and also, where appropriate, in their ongoing review and revision.
-
-

- SENIOR FELLOW
- SENIOR FELLOW(–FY2024)
- Akio Kurokawa
- Akio Kurokawa
- Areas of Expertise
-
- Public policy
- healthcare innovation policy
- science
- technology and innovation policy
- science for policy
-

Featured Content
-
Japan’s Population Decline and the Gijinkoku Visa’s Pathway to Settlement without Adequate Screening
Japan’s Population Decline and the Gijinkoku Visa’s Pathway to Settlement without Adequate Screening
-
Haiku: The Heart of Japan in 17 Syllables
Haiku: The Heart of Japan in 17 Syllables
-
In Japan, Men Have Complicated Views about Gender and Equality
In Japan, Men Have Complicated Views about Gender and Equality
-
Democracy in Crisis: Lessons from Japanese History
Democracy in Crisis: Lessons from Japanese History
-
Abalone
Abalone
















